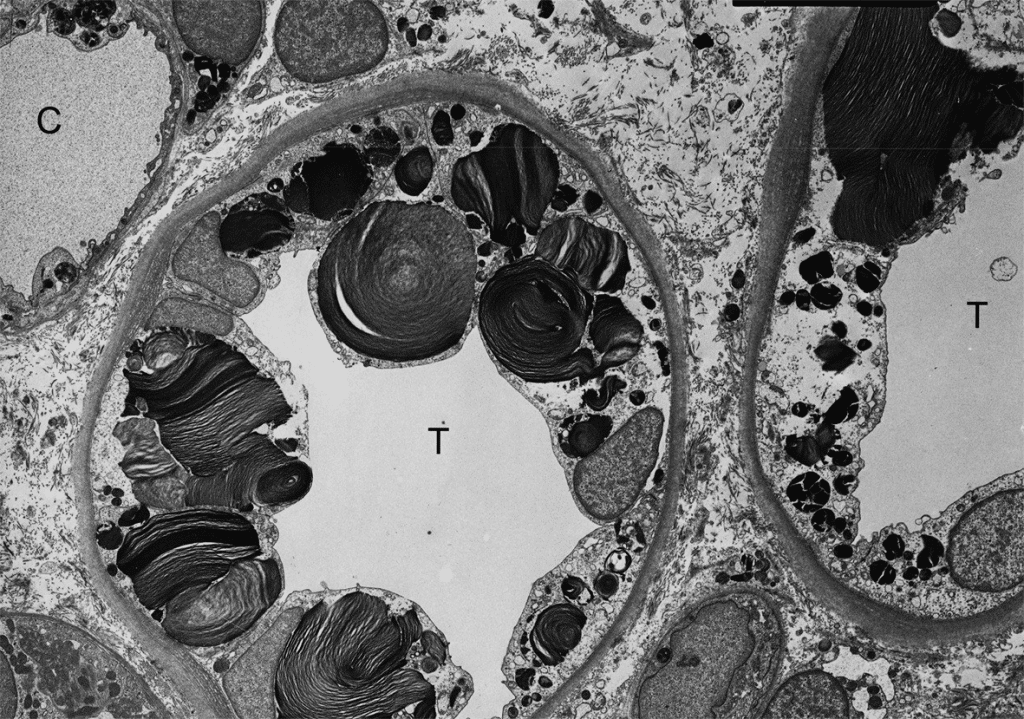
Болезнь Фабри, клинические признаки, возможности диагностики и лечения.
Болезнь Фабри, или болезнь Андерсона-Фабри (OMIM 301500; МКБ-10 E75,2) – наследственное заболевание из группы лизосомных болезней накопления, обусловленное значительным снижением или отсутствием фермента альфа-галактозидазы А. В результате происходит накопление гликофосфолипидов в лизосомах клеток различных органов: сердца, почек, нервной системы, эндотелия сосудов.
Болезнь Фабри наследуется по Х-сцепленному рецессивному типу наследования, для которого характерна передача матерью Х-хромосомы с патологической мутацией половине своих потомков. Мужчины имеют единственную Х-хромосому, что определяет классический вариант болезни, однако у части женщин, носительниц мутации гена альфа-галактозидазы А также развиваются тяжелые проявления болезни Фабри.
Частота болезни Фабри в популяции примерно 1-5:10 000.
Клинические признаки болезни Фабри могут быть разные у лиц мужского и женского пола и даже у членов одной семьи.
Основные проявления в детском возрасте − боли в кистях и стопах. ангиокератомы, гипогидроз, астения. В более старшем возрасте присоединяются боль в животе, поражение почек, сердца, ишемические атаки, инсульт. Заболевание носит прогрессирующий характер, смерть может наступать на 4 десятилетии жизни от сердечно-сосудистых, цереброваскулярных нарушений, почечной недостаточности.
У пациентов с болезнью Фабри в 70-80% случаев отмечаются выраженные изнуряющие невропатические боли в конечностях, ладонях, стопах, которые сопровождаются акропарестезиями. Характерны болевые кризы, приступы сильной жгучей боли, которые возникают уже с 2-х лет и провоцируются переменой погоды, лихорадкой, физической нагрузкой, могут длиться от нескольких секунд до нескольких недель не снимаются анальгетиками. Считается, что боль является результатом структурных повреждений нервных волокон в результате накопления субстрата.
Ангиокератомы, состоящие из скоплений эктатических кровеносных сосудов, от темно-красного до синюшного цвета обычно расположены в паховой области, на ягодицах, бедрах, постепенно с годами увеличиваются в количестве и размерах.
При болезни Фабри высок риск транзиторных ишемических атак и ишемических или геморрагических инсультов уже в возрасте от 20 до 50 лет, в том числе у каждого пятого в этой группе до 30 лет. Частота инсульта у больных болезнью Фабри составляет 6,9% у мужчин и 4,3% у женщин, инсульт нередко является дебютом болезни Фабри.
Первые симптомы поражения почек − микроальбуминурия и протеинурия появляются в возрасте 20-30 лет, которые с возрастом нарастают, отмечаются канальцевые нарушения. Постепенно снижается функция почек, что приводит к развитию терминальной почечной недостаточности, требующей заместительной почечной терапии или трансплантации почек и являющейся одной из причин смерти пациентов с болезнью Фабри.
Поражение сердца при болезни Фабри – наиболее распространенное и прогностически неблагоприятное проявление данного заболевания. Именно заболевания сердца стоят на первом месте среди причин смерти у пациентов с болезнью Фабри (34% среди мужчин и 57% среди женщин).
Характерным признаком поражения сердца является гипертрофия левого желудочка, которая выявляется с помощью ЭКГ, эхокардиографии и магнито-резонансной томографии. Утолщение стенок сердца связано не только с отложением гликосфинголипидов в миоцитах и проводящей ткани, но и с гипертрофией миоцитов и фиброзом, которые часто сопровождаются диастолической дисфункцией левого желудочка. Нарушения диастолической функции приводят к развитию сердечной недостаточности, которую выявляют у 11% мужчин и 6% женщин с болезнью Фабри. Часто встречаются желудочковые и предсердные аритмии, нарушения проводимости. Инфаркт миокарда диагностируют у 2% больных. Стенокардия наблюдается у 13-20% пациентов с болезнью Фабри чаще при наличии гипертрофии левого желудочка.
Для болезни Фабри характерны и офтальмологические нарушения, помутнение роговицы в виде завитков встречается у 70-90% больных, наблюдаются и задняя субкапсулярная катаракта и поражение сосудов сетчатки с тяжелой потерей зрения.
Изменения со стороны органов слуха и вестибулярного аппарата встречаются в виде прогрессирующего снижения слуха, шума в ушах, головокружения.
Заподозрить болезнь Фабри возможно при наличии системного заболевания с поражением почек, сердца, кожи, нервной системы, органа зрения. Однако поражение сердца или почек может быть единственным проявлением болезни Фабри. Предполагать болезнь Фабри следует у всех пациентов гипертрофией миокарда неясного происхождения и инсультом, развивающимся в молодом возрасте.
Учитывая наследственный характер заболевания, важным является и изучение семейного анамнеза, учитывая, что у родственников заболевание может быть нераспознанно.
При наличии любых перечисленных симптомов любой специалист направляет пациента для консультации в отдел медицинской генетики УЗ «Могилевский ОЛДЦ».
Врач-генетик проводит тщательный сбор анамнеза болезни, генетического анамнеза и с учетом жалоб пациента, результатов лабораторных, функциональных исследований при подозрении на болезнь Фабри назначает необходимые лабораторные диагностические тесты, позволяющие установить диагноз.
Проведение лабораторной диагностики проводится в ГУ РНПЦ «Мать и дитя» по биологическому материалу пациента, полученному с соблюдением необходимых условий по направлению врача-генетика.
С целью диагностики болезни Фабри определяют активность лизосомного фермента альфа-галактозидазы А в плазме или лейкоцитах крови, а также при исследовании высушенных капель крови на фильтровальной бумаге (такие образцы могут храниться несколько месяцев).
Снижение активности альфа-галактозидазы А является информативным показателем для диагностики болезни Фабри.
Молекулярно-генетические исследования гена альфа-галактозидазы А позволяют выявить различные мутации, являющиеся причиной болезни Фабри. Проведение данной лабораторной диагностики возможно в ГУ РНПЦ «Мать и дитя», материал для исследования.
Для лечения пациентов с болезнью Фабри проводится ферментозаместительная терапия рекомбинантными препаратами альфа-галактозидазы А. С этой целью применяют препараты агалсидазу альфа и агалсидазу бета.
Заместительная терапия уменьшает выраженность невропатической боли, вызывает регресс гипертрофии левого желудочка, стабилизацию функции почек, задерживает развитие почечной и сердечно-сосудистой недостаточности, улучшает качество жизни больных.
Так как заболевание имеет мультисистемный характер, в диагностике и лечении пациентов с болезнью Фабри для проведения симптоматической терапии необходимо участие врачей различных специальностей.
Врач-генетик
высшей квалификационной категории А.С.Бойша